bactopia
Tags: bacteria assembly annotation amr mlst genomics pipeline named-workflow
Comprehensive bacterial analysis pipeline for complete genomic characterization.
This workflow performs end-to-end analysis including quality control, assembly, annotation, antimicrobial resistance detection, MLST typing, and optional pathogen-specific analysis through Merlin. It processes raw sequencing reads and produces a complete genomic characterization suitable for downstream analysis.
Pipeline Overview
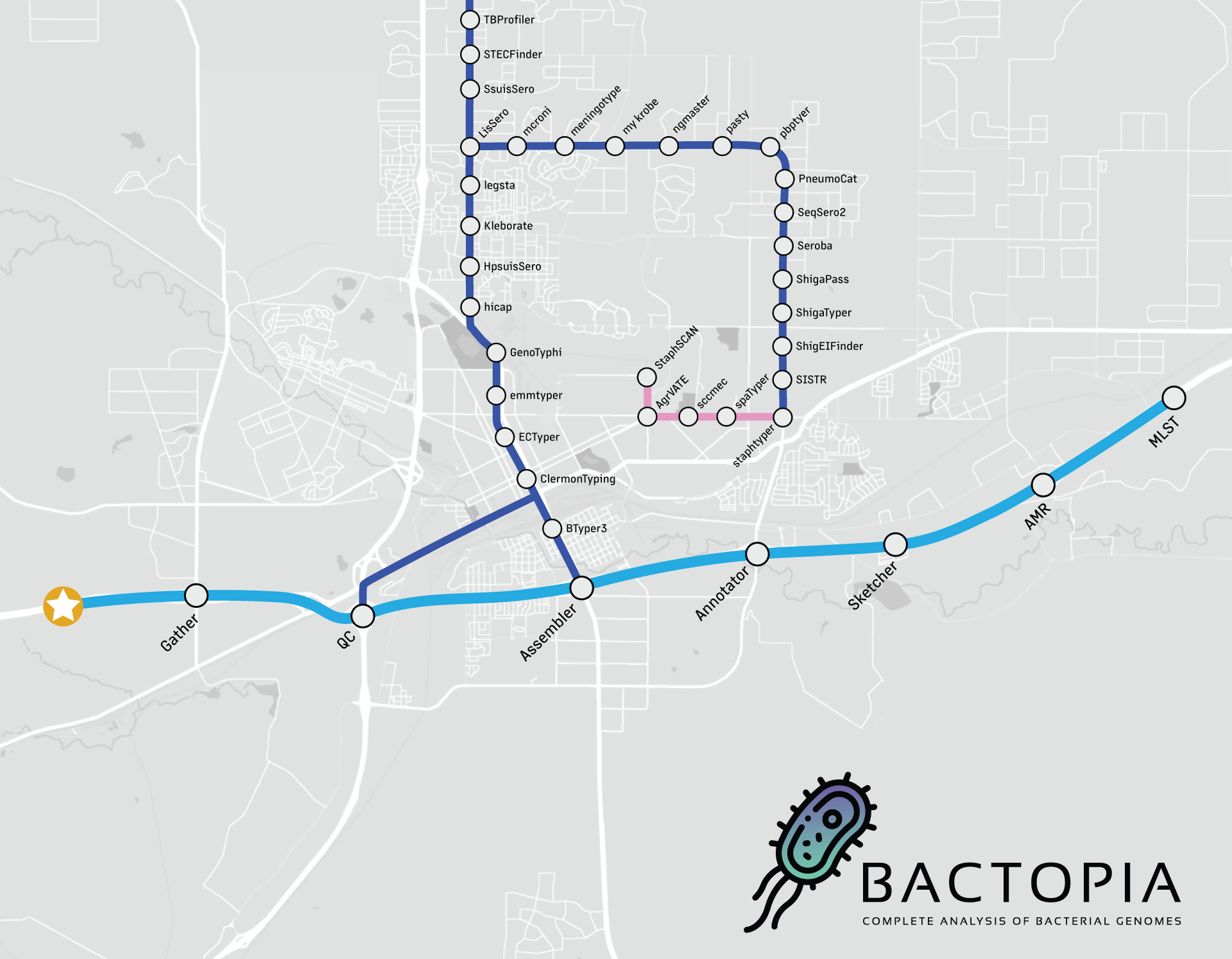
Looking at the workflow overview above, it might not look like much is happening, but I can assure you that a lot is going on. The workflow is broken down into 8 steps, which are:
- Gather - Collect all the data in one place
- QC - Quality control of the data
- Assembler - Assemble the reads into contigs
- Annotator - Annotate the contigs
- Sketcher - Create a sketch of the contigs, and query databases
- Sequence Typing - Determine the sequence type of the contigs
- Antibiotic Resistance - Determine the antibiotic resistance of the contigs and proteins
- Merlin - Automatically run species-specific tools based on distance
If you are looking for a guide to get started quickly, please check out the Beginner's Guide.
Step 1 - Gather
The main purpose of the gather step is to get all the samples into a single place. This
includes downloading samples from ENA/SRA or NCBI Assembly. The tools used are:
| Tool | Description |
|---|---|
| art | For simulating error-free reads for an input assembly |
| fastq-dl | Downloading FASTQ files from ENA/SRA |
| ncbi-genome-download | Downloading FASTA files from NCBI Assembly |
This gather step also does basic QC checks to help prevent downstream failures.
Failed Quality Checks
| Filename | Description |
|---|---|
| -gzip-error.txt | Sample failed Gzip checks and excluded from further analysis |
| -low-basepair-proportion-error.txt | Sample failed basepair proportion checks and excluded from further analysis |
| -low-read-count-error.txt | Sample failed read count checks and excluded from further analysis |
| -low-sequence-depth-error.txt | Sample failed sequenced basepair checks and excluded from further analysis |
Samples that fail any of the QC checks will be excluded from further analysis.
Those samples will generate a *-error.txt file with the error message. Excluding
these samples prevents downstream failures that cause the whole workflow to fail.
Example Error: Input FASTQ(s) failed Gzip checks
If input FASTQ(s) fail to pass Gzip test, the sample will be excluded from further analysis.
Example Text from <SAMPLE_NAME>-gzip-error.txt <SAMPLE_NAME> FASTQs failed Gzip tests. Please check the input FASTQs. Further analysis is discontinued.
Example Error: Input FASTQs have disproportionate number of reads
If input FASTQ(s) for a sample have disproportionately different number of reads
between the two pairs, the sample will be excluded from further analysis. You can
adjust this minimum read count using the --min_proportion parameter.
Example Text from <SAMPLE_NAME>-low-basepair-proportion-error.txt
<SAMPLE_NAME> FASTQs failed to meet the minimum shared basepairs. They
shared Y basepairs, with R1 having A bp and R2 having B bp. Further
analysis is discontinued.
Example Error: Input FASTQ(s) has too few reads
If input FASTQ(s) for a sample have less than the minimum required reads, the
sample will be excluded from further analysis. You can adjust this minimum read
count using the --min_reads parameter.
Example Text from <SAMPLE_NAME>-low-read-count-error.txt
<SAMPLE_NAME> FASTQ(s) contain X total reads. This does not exceed the required
minimum Y read count. Further analysis is discontinued.
Example Error: Input FASTQ(s) has too little sequenced basepairs
If input FASTQ(s) for a sample fails to meet the minimum number of sequenced
basepairs, the sample will be excluded from further analysis. You can
adjust this minimum read count using the --min_basepairs parameter.
Example Text from <SAMPLE_NAME>-low-sequence-depth-error.txt
<SAMPLE_NAME> FASTQ(s) contain X total basepairs. This does not exceed the
required minimum Y bp. Further analysis is discontinued.
Step 2 - QC
The qc module uses a variety of tools to perform quality control on Illumina and
Oxford Nanopore reads. The tools used are:
| Tool | Technology | Description |
|---|---|---|
| bbtools | Illumina | A suite of tools for manipulating reads |
| fastp | Illumina | A tool designed to provide fast all-in-one preprocessing for FastQ files |
| fastqc | Illumina | A quality control tool for high throughput sequence data |
| fastq_scan | Nanopore | A tool for quickly scanning FASTQ files |
| lighter | Illumina | A tool for correcting sequencing errors in Illumina reads |
| NanoPlot | Nanopore | A tool for plotting long read sequencing data |
| nanoq | Nanopore | A tool for calculating quality metrics for Oxford Nanopore reads |
| porechop | Nanopore | A tool for removing adapters from Oxford Nanopore reads |
| rasusa | Nanopore | Randomly subsample sequencing reads to a specified coverage |
Similar to the gather step, the qc step will also stop samples that fail to meet
basic QC checks from continuing downstream.
Failed Quality Checks
| Filename | Description |
|---|---|
| .error-fastq.gz | A gzipped FASTQ file of reads that failed QC |
| -low-read-count-error.txt | Sample failed read count checks and excluded from further analysis |
| -low-sequence-coverage-error.txt | Sample failed sequenced coverage checks and excluded from further analysis |
| -low-sequence-depth-error.txt | Sample failed sequenced basepair checks and excluded from further analysis |
Samples that fail any of the QC checks will be excluded from further analysis.
Those samples will generate a *-error.txt file with the error message. Excluding
these samples prevents downstream failures that cause the whole workflow to fail.
Example Error: After QC, too few reads remain
If after cleaning reads, a sample has less than the minimum required reads, the
sample will be excluded from further analysis. You can adjust this minimum read
count using the --min_reads parameter.
Example Text from <SAMPLE_NAME>-low-read-count-error.txt
<SAMPLE_NAME> FASTQ(s) contain X total reads. This does not exceed the required
minimum Y read count. Further analysis is discontinued.
Example Error: After QC, too little sequence coverage remains
If after cleaning reads, a sample has failed to meet the minimum sequence
coverage required, the sample will be excluded from further analysis. You can
adjust this minimum read count using the --min_coverage parameter.
Note: This check is only performed when a genome size is available.
Example Text from <SAMPLE_NAME>-low-sequence-coverage-error.txt
After QC, <SAMPLE_NAME> FASTQ(s) contain X total basepairs. This does not
exceed the required minimum Y bp (Zx coverage). Further analysis is
discontinued.
Example Error: After QC, too little sequenced basepairs remain
If after cleaning reads, a sample has failed to meet the minimum number of
sequenced basepairs, the sample will be excluded from further analysis. You can
adjust this minimum read count using the --min_basepairs parameter.
Example Text from <SAMPLE_NAME>-low-sequence-depth-error.txt
<SAMPLE_NAME> FASTQ(s) contain X total basepairs. This does not exceed the
required minimum Y bp. Further analysis is discontinued.
Step 3 - Assembler
The assembler module uses a variety of assembly tools to create an assembly of
Illumina and Oxford Nanopore reads. The tools used are:
| Tool | Description |
|---|---|
| Dragonflye | Assembly of Oxford Nanopore reads, as well as hybrid assembly with short-read polishing |
| Shovill | Assembly of Illumina paired-end reads |
| Shovill-SE | Assembly of Illumina single-end reads |
| Unicycler | Hybrid assembly, using short-reads first then long-reads |
Summary statistics for each assembly are generated using assembly-scan.
--short_polish over --hybrid with recent ONT sequencingUsing Unicycler (--hybrid) to create a hybrid
assembly works great when you have low-coverage noisy long-reads. However, if you are
using recent ONT sequencing, you likely have high-coverage and using the --short_polish
method is going to yield better results (and be faster!) than --hybrid.
Failed Quality Checks
| Filename | Description |
|---|---|
| -assembly-error.txt | Sample failed assembly checks and excluded from further analysis |
Samples that fail any of the QC checks will be excluded from further analysis.
Those samples will generate a *-error.txt file with the error message. Excluding
these samples prevents downstream failures that cause the whole workflow to fail.
Example Error: Assembled Successfully, but 0 Contigs
If a sample assembles successfully, but 0 contigs are formed, the sample will be excluded from further analysis.
Example Text from <SAMPLE_NAME>-assembly-error.txt <SAMPLE_NAME> assembled successfully, but 0 contigs were formed. Please investigate <SAMPLE_NAME> to determine a cause (e.g. metagenomic, contaminants, etc...) for this outcome. Further assembly-based analysis of <SAMPLE_NAME> will be discontinued.
Example Error: Assembled successfully, but poor assembly size
If your sample assembles successfully, but the assembly size is less than the minimum
allowed genome size, the sample will be excluded from further analysis. You can
adjust this minimum size using the --min_genome_size parameter.
Example Text from <SAMPLE_NAME>-assembly-error.txt
<SAMPLE_NAME> assembled size (000 bp) is less than the minimum allowed genome
size (000 bp). If this is unexpected, please investigate <SAMPLE_NAME> to
determine a cause (e.g. metagenomic, contaminants, etc...) for the poor assembly.
Otherwise, adjust the --min_genome_size parameter to fit your need. Further
assembly based analysis of <SAMPLE_NAME> will be discontinued.
Step 4 - Annotator
The annotator step uses either Prokka (default)
or Bakta (via --use_bakta) to annotate
assembled contigs with functional information including genes, proteins, rRNA, tRNA,
and other genomic features.
Step 5 - Sketcher
The sketcher module uses Mash and
Sourmash to create sketches and query
RefSeq and GTDB.
Step 6 - Sequence Typing
The mlst step uses mlst to scan assemblies against
PubMLST typing schemes and determine the sequence type.
Step 7 - Antibiotic Resistance
The amrfinderplus step uses AMRFinder+ to identify
antimicrobial resistance genes and point mutations from both assembled contigs and
annotated protein sequences.
Step 8 - Merlin
The merlin step automatically selects and runs species-specific typing tools based on
Mash distance results from the sketcher step. Enable with --ask_merlin. See the
Pathogen-Specific Analysis output section below for the
full list of supported organisms and tools.
Usage
Bactopia CLI:
bactopia \
--input samples.csv \
--outdir results/
Nextflow:
nextflow run bactopia/bactopia \
--input samples.csv \
--outdir results/
Outputs
Expected Output Files
<BACTOPIA_DIR>
├── <SAMPLE_NAME>
│ ├── main
│ │ ├── annotator
│ │ │ └── prokka
│ │ │ ├── <SAMPLE_NAME>-blastdb.tar.gz
│ │ │ ├── <SAMPLE_NAME>.faa.gz
│ │ │ ├── <SAMPLE_NAME>.ffn.gz
│ │ │ ├── <SAMPLE_NAME>.fna.gz
│ │ │ ├── <SAMPLE_NAME>.fsa.gz
│ │ │ ├── <SAMPLE_NAME>.gbk.gz
│ │ │ ├── <SAMPLE_NAME>.gff.gz
│ │ │ ├── <SAMPLE_NAME>.sqn.gz
│ │ │ ├── <SAMPLE_NAME>.tbl.gz
│ │ │ ├── <SAMPLE_NAME>.tsv
│ │ │ ├── <SAMPLE_NAME>.txt
│ │ │ └── logs
│ │ │ ├── <SAMPLE_NAME>.err
│ │ │ ├── <SAMPLE_NAME>.log
│ │ │ ├── nf.command.{begin,err,log,out,run,sh,trace}
│ │ │ └── versions.yml
│ │ ├── assembler
│ │ │ ├── <SAMPLE_NAME>.fna.gz
│ │ │ ├── <SAMPLE_NAME>.tsv
│ │ │ ├── logs
│ │ │ │ ├── nf.command.{begin,err,log,out,run,sh,trace}
│ │ │ │ ├── shovill-se.log
│ │ │ │ └── versions.yml
│ │ │ └── supplemental
│ │ │ └── shovill.corrections
│ │ ├── gather
│ │ │ ├── <SAMPLE_NAME>-meta.tsv
│ │ │ └── logs
│ │ │ ├── nf.command.{begin,err,log,out,run,sh,trace}
│ │ │ └── versions.yml
│ │ ├── qc
│ │ │ ├── <SAMPLE_NAME>_SE.fastq.gz
│ │ │ ├── logs
│ │ │ │ ├── <SAMPLE_NAME>-fastp.log
│ │ │ │ ├── nf.command.{begin,err,log,out,run,sh,trace}
│ │ │ │ └── versions.yml
│ │ │ └── supplemental
│ │ │ ├── <SAMPLE_NAME>.fastp.html
│ │ │ ├── <SAMPLE_NAME>.fastp.json
│ │ │ ├── <SAMPLE_NAME>_SE-final.json
│ │ │ ├── <SAMPLE_NAME>_SE-final_fastqc.html
│ │ │ ├── <SAMPLE_NAME>_SE-final_fastqc.zip
│ │ │ ├── <SAMPLE_NAME>_SE-original.json
│ │ │ ├── <SAMPLE_NAME>_SE-original_fastqc.html
│ │ │ └── <SAMPLE_NAME>_SE-original_fastqc.zip
│ │ └── sketcher
│ │ ├── <SAMPLE_NAME>-k21.msh
│ │ ├── <SAMPLE_NAME>-k31.msh
│ │ ├── <SAMPLE_NAME>-mash-refseq88-k21.txt
│ │ ├── <SAMPLE_NAME>-sourmash-gtdb-rs207-k31.txt
│ │ ├── <SAMPLE_NAME>.sig
│ │ └── logs
│ │ ├── nf.command.{begin,err,log,out,run,sh,trace}
│ │ └── versions.yml
│ └── tools
│ ├── amrfinderplus
│ │ ├── <SAMPLE_NAME>.tsv
│ │ └── logs
│ │ ├── nf.command.{begin,err,log,out,run,sh,trace}
│ │ └── versions.yml
│ └── mlst
│ ├── <SAMPLE_NAME>.tsv
│ └── logs
│ ├── nf.command.{begin,err,log,out,run,sh,trace}
│ └── versions.yml
└── bactopia-runs
└── bactopia-<TIMESTAMP>
├── merged-results
│ ├── amrfinderplus.tsv
│ ├── assembly-scan.tsv
│ ├── logs
│ │ ├── amrfinderplus-concat
│ │ │ ├── nf.command.{begin,err,log,out,run,sh,trace}
│ │ │ └── versions.yml
│ │ ├── assembly-scan-concat
│ │ │ ├── nf.command.{begin,err,log,out,run,sh,trace}
│ │ │ └── versions.yml
│ │ ├── meta-concat
│ │ │ ├── nf.command.{begin,err,log,out,run,sh,trace}
│ │ │ └── versions.yml
│ │ └── mlst-concat
│ │ ├── nf.command.{begin,err,log,out,run,sh,trace}
│ │ └── versions.yml
│ ├── meta.tsv
│ └── mlst.tsv
└── nf-reports
├── bactopia-dag.dot
├── bactopia-report.html
└── bactopia-timeline.html
Quality Control
| File | Description |
|---|---|
supplemental/*_fastqc.* | FastQC quality control reports for raw and cleaned reads |
supplemental/*-NanoPlot.* | NanoPlot reports for Nanopore reads |
supplemental/*.fastp.* | Fastp quality reports (when applicable) |
supplemental/*_original.json | Quality metrics for original reads |
supplemental/*_final.json | Quality metrics for final reads |
Assembly
| File | Description |
|---|---|
*.fasta | Assembled genome sequences |
assembly-stats.tsv | Assembly quality metrics |
merged-assembly-stats.tsv | Consolidated assembly statistics |
Annotation
Output format depends on chosen annotation tool (Bakta or Prokka)
| File | Description |
|---|---|
*.gff.gz | Genome annotation in GFF3 format (compressed) |
*.gbk.gz | Genome annotation in GenBank format (compressed) |
*.faa.gz | Protein sequences (compressed) |
*.fna.gz | Nucleotide sequences from annotation (compressed) |
*.ffn.gz | Feature nucleotide sequences (compressed) |
annotation.tsv | Annotation summary tables |
blastdb.* | BLAST database created from annotation |
Typing
| File | Description |
|---|---|
mlst.tsv | MLST sequence type results |
merged-mlst.tsv | Consolidated MLST results |
Antimicrobial Resistance
| File | Description |
|---|---|
amrfinderplus.tsv | AMR gene detection results |
amrfinderplus.mutation.tsv | AMR point mutation results |
merged-amrfinderplus.tsv | Consolidated AMR results |
Comparative Analysis
| File | Description |
|---|---|
*-k21.msh | Mash sketch files (k=21) |
*-k31.msh | Mash sketch files (k=31) |
*-mash-refseq88-*.txt | Mash screening results against RefSeq |
*.sig | Sourmash signatures |
sourmash-*.txt | Sourmash classification results |
Pathogen-Specific Analysis
Only created if --ask_merlin is enabled
| File | Description |
|---|---|
merlin/clermontyping/* | E. coli phylogroup typing |
merlin/ectyper/* | Enterotoxigenic E. coli typing |
merlin/shigatyper/* | Shigella serotype prediction |
merlin/shigapass/* | Shigella passive surveillance |
merlin/shigeifinder/* | Shigella and EIEC detection |
merlin/stecfinder/* | STEC detection and typing |
merlin/emmtyper/* | S. pyogenes emm typing |
merlin/hicap/* | H. influenzae capsular typing |
merlin/hpsuissero/* | H. parasuis serotyping |
merlin/kleborate/* | Klebsiella species typing |
merlin/staphtyper/* | S. aureus spa typing |
merlin/agrvate/* | S. aureus agr typing |
merlin/sccmec/* | S. aureus SCCmec typing |
Merged Results
Run-level aggregated results from all samples
| File | Description |
|---|---|
samplesheet.tsv | Sample metadata and quality metrics |
Audit Trail
Below are files that can assist you in understanding which parameters and program versions were used.
Logs
Each process that is executed will have a folder named logs. In this folder are helpful
files for you to review if the need ever arises.
| Extension | Description |
|---|---|
| .begin | An empty file used to designate the process started |
| .err | Contains STDERR outputs from the process |
| .log | Contains both STDERR and STDOUT outputs from the process |
| .out | Contains STDOUT outputs from the process |
| .run | The script Nextflow uses to stage/unstage files and queue processes based on given profile |
| .sh | The script executed by bash for the process |
| .trace | The Nextflow trace report for the process |
| versions.yml | A YAML formatted file with program versions |
Nextflow Reports
These Nextflow reports provide great a great summary of your run. These can be used to optimize resource usage and estimate expected costs if using cloud platforms.
| Filename | Description |
|---|---|
| bactopia-dag.dot | The Nextflow DAG visualization |
| bactopia-report.html | The Nextflow Execution Report |
| bactopia-timeline.html | The Nextflow Timeline Report |
| bactopia-trace.txt | The Nextflow Trace report |
Parameters
Required Parameters
The following parameters are how you will provide either local or remote samples to be processed by Bactopia.
| Parameter | Type | Default | Description |
|---|---|---|---|
--samples | string | A FOFN (via bactopia prepare) with sample names and paths to FASTQ/FASTAs to process | |
--r1 | string | First set of compressed (gzip) Illumina paired-end FASTQ reads (requires --r2 and --sample) | |
--r2 | string | Second set of compressed (gzip) Illumina paired-end FASTQ reads (requires --r1 and --sample) | |
--se | string | Compressed (gzip) Illumina single-end FASTQ reads (requires --sample) | |
--ont | string | Compressed (gzip) Oxford Nanopore FASTQ reads (requires --sample) | |
--hybrid | boolean | false | Create hybrid assembly using Unicycler. (requires --r1, --r2, --ont and --sample) |
--short_polish | boolean | false | Create hybrid assembly from long-read assembly and short read polishing. (requires --r1, --r2, --ont and --sample) |
--sample | string | Sample name to use for the input sequences | |
--accessions | string | A file containing ENA/SRA Experiment accessions or NCBI Assembly accessions to processed | |
--accession | string | Sample name to use for the input sequences | |
--assembly | string | A assembled genome in compressed FASTA format. (requires --sample) | |
--check_samples | boolean | false | Validate the input FOFN provided by --samples |
Dataset Parameters
Define where the pipeline should find input data and save output data.
| Parameter | Type | Default | Description |
|---|---|---|---|
--species | string | Name of species for species-specific dataset to use | |
--ask_merlin | boolean | Ask Merlin to execute species specific Bactopia tools based on Mash distances | |
--coverage | integer | 100 | Reduce samples to a given coverage, requires a genome size |
--genome_size | integer | 0 | Expected genome size (bp) for all samples, required for read error correction and read subsampling |
--use_bakta | boolean | Use Bakta for annotation, instead of Prokka |
Optional Parameters
These optional parameters can be useful in certain settings.
| Parameter | Type | Default | Description |
|---|---|---|---|
--outdir | string | bactopia | Base directory to write results to |
Nextflow Profile Parameters
Parameters to fine-tune your Nextflow setup.
| Parameter | Type | Default | Description |
|---|---|---|---|
--datasets_cache | string | <HOME>/.bactopia/datasets | Directory where downloaded datasets should be stored. |
Helpful Parameters
Uncommonly used parameters that might be useful.
| Parameter | Type | Default | Description |
|---|---|---|---|
--wf | string | bactopia | Specify which workflow or Bactopia Tool to execute |
--list_wfs | boolean | List the available workflows and Bactopia Tools to use with '--wf' | |
--help_all | boolean | An alias for --help --show_hidden_params | |
--version | boolean | Display version text. |
Composition
This workflow uses the following subworkflows:
- amrfinderplus - Find antimicrobial resistance genes and point mutations.
- bactopia_assembler - Assemble bacterial genomes using automated assembler selection.
- bactopia_datasets - Download and provide pre-compiled datasets required by Bactopia.
- bactopia_gather - Search, validate, gather, and standardize input samples.
- bactopia_qc - Perform comprehensive quality control on sequencing reads.
- bactopia_sketcher - Create genomic sketches and perform rapid taxonomic classification.
- bakta - Rapid bacterial genome annotation.
- merlin - MinER assisted species-specific bactopia tool seLectIoN.
- mlst - Determine multilocus sequence types (MLST) from bacterial assemblies.
- prokka - Annotate bacterial genomes with functional information.